

- 3.65 MB
- 2022-04-29 14:39:18 发布


- 1、本文档共5页,可阅读全部内容。
- 2、本文档内容版权归属内容提供方,所产生的收益全部归内容提供方所有。如果您对本文有版权争议,可选择认领,认领后既往收益都归您。
- 3、本文档由用户上传,本站不保证质量和数量令人满意,可能有诸多瑕疵,付费之前,请仔细先通过免费阅读内容等途径辨别内容交易风险。如存在严重挂羊头卖狗肉之情形,可联系本站下载客服投诉处理。
- 文档侵权举报电话:19940600175。
'红外分光光度法
概述红外区:0.76~500μm(或1000μm)近红外区:0.76~2.5μm13158~4000σcm-1中红外区:2.5~50μm4000~200σcm-1远红外区:50~500μm200~20σcm-1红外分光光度法:利用物质对红外光区电磁辐射的选择性吸收的特性进行结构分析、定性、定量分析方法。红外光谱的表示方法:T%~σ;T%~λ
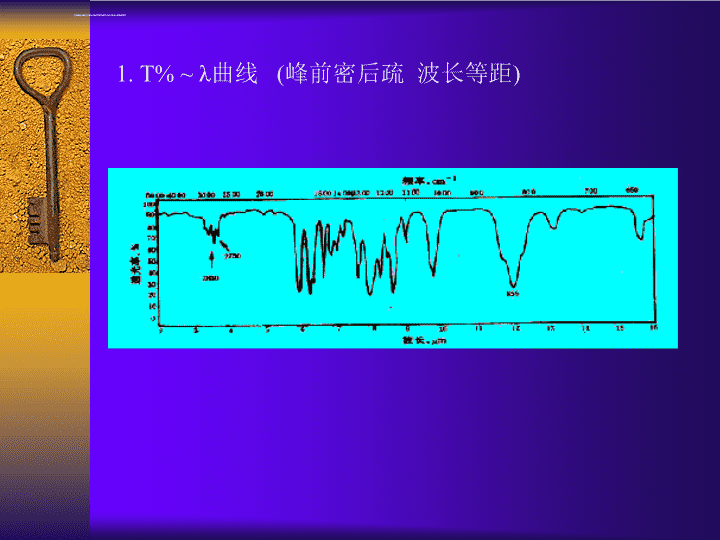
1.T%~λ曲线(峰前密后疏波长等距)
2.T%~曲线(峰前疏后密波数等距)
红外吸收光谱与紫外吸收光谱的区别1.起源不同UV-电子光谱IR-振转光谱2.适用范围广UV:研究芳香族或具有共轭结构的不饱和脂肪族化合物及某些无机物,不适用饱和有机化合物。IR:所有红外吸收的有机化合物3.测定样品物态UV:溶液、少数蒸气IR:气、液、固4.特征性强5.用途:鉴定化合物类别、鉴定官能团、推测化合物的分子结构式(IR)定量分析,推测有机化合物共轭骨架(UV)
基本原理红外吸收曲线:用峰位、峰强、峰数描述吸收峰产生原因、峰位、峰强、峰数,及影响因素:一、振动能级与振动光谱1.振动能级分子振动能级差0.05~1.0ev转动能级差0.0001~0.025ev。因此,分子发生振动能级跃迁必然同时伴随转动能级跃迁,无法测纯振动光谱。若把分子A-B的两个原子视为两个小球,则两个原子间的伸缩振动可近似看成沿键轴方向的简谐振动,双原子可视为谐振子。
二、振动形式双原子分子---伸缩多原子分子---伸缩+弯曲1.伸缩振动:s,as例:-CH2,-CH3~2850cm-1~2925cm-1,~2870cm-1~2960cm-1除-CH2,-CH3外,凡含有两个或两个以上相同键的基团也都有对称和反对称两种伸缩振动形式。2.弯曲振动:分为(1)面内弯曲振动----在由几个原子构成的平面内进行。面内剪式弯曲---键角规律性变化,似剪刀“开”“闭”。例:-CH2(CH)1465±20cm-1
面内摇摆振动---基团作为一个整体在平面内摇摆,键角无变化。例:-CH2(CH)~720cm-1_(CH2)n-n4(2)面外弯曲振动---弯曲振动在垂直于由几个原子构成的平面内进行。面外摇摆振动---两个原子同时垂直向面上或面下形成的振动。例:-CH2CH2~1300cm-1锩曲振动---一个原子垂直向面上,另一个原子垂直向面下例:-CH2CH2~1250cm-1(3)变形振动----多个化学键端的原子相对于分子其余部分的弯曲振动。
对称变形振动s---以-CH3为例,在振动过程中,三个C-H键与轴线组成的夹角对称的缩小或扩大,形似花瓣开、闭。例:-CH3sCH3~1375cm-1不对称变形振动as---在振动过程中,二个角缩小,一个角增大,或相反。(交替变化)例:-CH3asCH3~1460cm-1以上讨论了各种振动形式,一般地,每种形式的振动,都能产生振动能级跃迁,产生相应的吸收峰,但也有例外。
伸缩振动
弯曲振动
振动自由度即分子独立振动的数目。双原子分子---伸缩振动多原子分子---伸缩、弯曲(复杂)在中红外区,分子的运动形式有三种:平动(平移)、振动、转动如是非线型分子,除平动外,整个分子还绕三个坐标轴转动,因而,还有三个转动自由度,从总数扣除3+3,剩下3N-6个振动自由度。
如是线型分子,转动自由度为2(以键轴为轴的转动惯量=0,不发生能量变化),则振动自由度为3N-5由振动自由度可估计基频峰的可能数目。例1:非线型分子----H2O振动自由度=33-6=3说明水分子有三种基本振动形式,可能产生三个吸收峰。即3652cm-13756cm-11595cm-1例2:线型分子---CO2振动自由度=33-5=4说明CO2分子有四种振动形式,理论上产生四个吸收峰,但实际产生2个,原因?
CO2分子的四种振动形式:+-+O=C=OO=C=O后二种振动形式虽不同,但振动频率相等,基频峰在图谱同一位置出现,合并为一个峰,这种现象称为简并。简并是使基频峰数小于振动自由度的原因之一。
红外吸收光谱产生的条件和吸收峰强度一、红外吸收光谱的产生必须满足下列条件:1.红外辐射的能量必须与分子的振动能级差相等,即:hL=EV或EL=V·h或L=V·2.分子振动过程中其偶极矩必须发生变化,即0,只有红外活性振动才能产生吸收峰。总结:分子吸收红外线发生能级跃迁,产生基频峰必须满足以上二个条件,缺一不可。判断某振动是否是红外活性振动,看分子的对称性。例:CO2为对称分子,为非活性振动,无峰。
(二)吸收峰的强度红外光谱图中吸收峰的高、矮可说明峰的相对强度。峰的绝对强度用摩尔吸光系数描述谱带强度的划分>100非常强峰VS20~100强峰S10~20中强峰m1~10弱峰W<1非常弱峰VW
吸收峰的位置峰位或称谱带位置,即振动能级跃迁时所吸收红外线的波长(波数)。(一)基频峰和泛频峰1.基频峰是分子吸收一定频率的红外线,振动能级由基态(V=0)跃迁至第一激发态(V=1),所产生的吸收峰。根据L=V·(分子或基团的振动频率)V=1L=2.泛频峰真实分子的振动不是简谐振动(b~b´).量子力学证明,非谐性振子的总能量应为:X为非谐性常数,表示分子振动的非谐性大小。非谐性振子的选律是V=±1、±2、±3…,亦即不仅可在相邻能级间跃迁,而且可一次跃迁两个或更多能级。
若由(V=0)(V=2),V=2L=2所产生的吸收峰称为二倍频峰。(类推)---统称倍频峰。二倍频峰较弱但仍能看到,三倍…因跃迁几率很小,常观测不到。由于分子振动的非谐振性质,位能曲线中的能级差不等距,随V增大,E逐渐减小。所以倍频峰的频率不是基频峰频率的整数倍,而是略小。除倍频峰外,有些弱峰由两个或多个基频峰频率的和或差产生,1+2…峰称为合频峰,1+2…差频峰。倍频峰、合频峰、差频峰统称为泛频峰。泛频峰很弱,不易辨认,光谱复杂,但增加光谱的特征性。例:取代苯的泛频峰2000~1667cm-1(5~6m),为苯环上=CH的倍频峰,特征性很强。
影响峰位的因素1.分子内部结构因素(1)电子效应诱导效应---吸电子基团的诱导效应使吸收峰向高频方向移动。例:共轭效应---使吸收峰向低频方向移动。例:
(2)空间效应环张力效应---当环有张力时,环内双键被消弱,其伸缩振动频率降低;而环外双键被增强,其伸缩振动频率增加,峰强也增加。例:氢键---使伸缩振动频率降低。分子内氢键---对峰位影响极明显,但不受浓度的影响,有助于结构分析。
例:2-羟基-4-甲氧基苯乙酮因分子内形成氢键,使羰基和羟基的伸缩振动频率大幅度降低。通常:O-H:3705~3200cm-1(酚羟基)C=O:1700~1670cm-1(芳酮)形成氢键后:O-H:2835cm-1(酚羟基)C=O:1623cm-1(芳酮)分子间氢键---受浓度影响较大,随浓度变化峰位发生变化,可利用稀释法判断分子间氢键。
例:醇与酚的羟基,在极稀溶液中呈游离状态。随浓度增加分子间形成氢键而成二聚体或多聚体。使O-H降低。游离(3640cm-1)二聚体(3515cm-1)多聚体(3350cm-1)另外,还有互变异构、费米共振等
2.外部因素(1)物态效应:同一化合物不同的聚集状态,其吸收频率和强度都会发生变化。例:正己酸在液态和气态的红外图谱
低压下的气体,分子间作用力极小,可得到孤立分子的窄峰,增大压力,峰变宽。液态图谱由于分子间作用力存在而变宽,如出现缔合或分子内氢键时,其吸收峰的频率、数目、强度均可能发生重大变化。固态图谱的峰比液态的峰更尖锐且更多,测定固态红外图谱用于鉴定最可靠。如化合物有几种晶型,各种晶型的图谱也不同。(2)溶剂效应:在极性溶剂中,溶质的极性基团(C=O、-N=O)的伸缩振动频率随溶剂极性的增加而降低(峰右移),而峰强往往增加。因形成氢键。为避免上述影响,在测定溶液的光谱时,尽可能在非极性稀溶液中测定。
特征区和指纹区为便于解析,通常把中红外区划分为二个区域。1.特征区4000~1300(4000~1250)cm-12.指纹区1300~400(1250~400)cm-1
特征峰和相关峰(一)特征峰定义:可用于鉴别官能团存在的吸收峰。对于多原子分子,详细分析各种振动形式很困难,对于吸收峰的识别,是通过对比大量图谱总结规律,然后从理论上得到证明。例:1700左右(羰基)、2240(腈基)。
(二)相关峰由一个官能团产生的一组相互依存的吸收峰。在多原子分子中,一个官能团可能有数种振动形式,而每种红外活性振动,一般均能产生相应吸收峰,有时还能看到泛频峰。故一个官能团会产生一组相关峰。用一组相关峰确定一个官能团的存在,是光谱解析的一条重要原则。如光谱中不存在某官能团的特征峰,也可否定某官能团存在。
有机化合物的典型光谱讨论典型光谱,可熟悉各种官能团的特征峰和相关峰及其与分子结构的关系,便于解析,进行结构分析。一、脂肪烃类1.烷烃主要特征峰有:C-H:-CH3as2962±10cm-1(S)s2872±10cm-1(S)-CH2as2926±10cm-1(S)s2853±10cm-1(S)-CH2890±10cm-1(m)常被掩盖C-H:-CH3as1450±20cm-1(S)s1380~1370cm-1(S)-CH21465±20cm-1(m)~722cm-1(m)(n4)
2.烯烃R-CH=CH2主要特征峰=CH(3100~3000cm-1)(m)C=C(1650cm-1)=CH(1000~650cm-1)3.炔烃R-CCH主要特征峰:CHCC3333~3267cm-12260~2100cm-1
二、芳香烃类一组相关峰1.H3100~3000cm-1(W~m,通常有几个峰)2.泛频峰2000~1667cm-1(vw)3.苯环骨架振动C=C1650~1430cm-1(双峰),~1600cm-1(w);~1500cm-1(s)4.芳氢面内弯曲振动H1250~1000cm-1(w)5.芳氢面外弯曲振动H910~665cm-1(s)由于泛频峰很弱,H常与该区域其它峰重叠不易辨认,故1、3、5为主要特征峰。
三、醇、酚、醚1.醇和酚R-OHAr-OH都有OH、C-O、OH(但OH的特征性差)区别:酚有芳环特征。特点:OH:游离的醇或酚3640~3610cm-1(S,尖)二聚体3600~3500(S,稍宽)多聚体3500~3200(S,宽)游离的OH峰及形成分子内氢键的OH峰较尖锐,形成分子间氢键较宽,并随浓度的增加向低波数移动。C-O1260~1000(S)饱和伯醇1085~1050cm-1饱和仲醇1124~1087饱和叔醇1205~1124OH1420~1330cm-1
2.醚R-O-R特点:无OH峰,特征峰:SC-O-C
四、羰基化合物酮、醛、酸、酯、酰卤、酸酐、酰胺和内酰胺特点:均含C=O、强度大、干扰少、最易识别。(一)酮R-COR特征峰:C=O1715cm-1(基准值)改变羰基周围环境,峰位变化(共轭1685~1665)环酮随环张力增加频率升高。(二)醛R-COH特征峰:C=O1725cm-1(S)CH(O)~2820、~2720cm-1C=O峰位与结构的关系与酮类似。(三)酰氯R-COCl特征峰:C=O1800cm-1(S,基准值),共轭右移。
(三)羧酸R-COOH主要特征峰:OH3400~2500cm-1(固态或液态,宽,中心在3000附近,与其它重叠,淹没,露峰尖)C=O1740~1650cm-1OH955~915cm-1(四)酯R-C00R´主要特征峰:C=O1735cm-1(S)C-O-C1300~1000cm-1(五)酸酐R-CO-O-OCR主要特征峰:C=O双峰1850~1800cm-1(S)1780~1740cm-1(S)C-O1300~900cm-1(S)
五、含氮化合物(一)酰胺类R-CO-NH2主要特征峰:NH3500~3100cm-1(S)C=O1680~1630cm-1(S)NH1640~1550cm-1特点:NH伯酰胺为双峰仲酰胺为单峰NH3270cm-1锐峰叔酰胺无此峰酰胺的C=O及NH峰分别称为酰胺谱带Ⅰ及谱带Ⅱ,是酰胺的较主要特征峰。环酰胺的C=O频率与环张力有关,环张力增大,C=O频率增大。
(二)胺类化合物R-NH2主要特征峰:NH3500~3300cm-1伯胺双峰,仲胺单峰,叔胺无此峰NH伯胺1650~1590,仲胺1650~1550cm-1CN1360~1020cm-1(m)NH900~650cm-1(S)
(三)硝基化合物R-NO2主要特征峰:asNO21600~1500cm-1(S);sNO21390~1300cm-1(S)强度很大,很容易辨认。在芳香族硝基化合物中,由于硝基的存在,使苯环的特征峰明显减弱。
红外分光光度计主要部件:光源吸收池(或固体样品装置)单色器检测器记录装置(显示装置,数据处理及储存系统)1.辐射源(光源)硅碳棒(Globar):碳化硅制成中间细两头粗的实心棒,中间发光,直径5mm,长5cm,工作温度1200℃,寿命长、发光面积大。Nernst灯:由稀有金属氧化物的混合物烧结而成。工作温度1750℃。最大发射波数7100cm-1.特殊线圈(Specialcoil):特殊金属丝制成,通电热炽产生红外线。2.色散元件多用反射光栅3.检测器真空热电偶Golay池(气胀式检测器)4.吸收池
干涉分光型红外分光光度计(FT-IR)1.工作原理FT-IR仪器的工作原理与色散型仪器的原理有很大不同,主要表现在单色器的差别。常用单色器为Michelson干涉仪。由光源发出的红外辐射,通过干涉仪产生干涉图,通过样品后,得到带有样品信息的干涉图。用计算机解出干涉图函数的Fourier余璇变换,即得到样品的红外光谱。2.检测器由于FT-IR的全程扫描<1秒,一般检测器的响应时间不能满足要求。多用热电型硫酸三甘肽(TGS)或光电导型如汞镉碲(MCT)检测器,这些检测器的响应时间为1微秒。(光源、吸收池等部件与色散型仪器通用)
制样气、液及固态样品均可测红外光谱,固态样品最方便。对样品的主要要求:1.样品的纯度要>98%,以便与纯化合物光谱对照(Sadtler)。2.样品应不含水分。(一)固态样品制样方法三种压片法:用200目光谱纯、干燥的KBr压片。糊剂法(软膏法):固体样品研细,滴加液体石蜡或全氟代烃,研成糊剂。夹于可拆卸池的两块窗片中或空白KBr片中。薄膜法:将固体样品溶于挥发性溶剂中,涂于窗片或空白KBr片上,溶剂挥发后,样品遗留而成薄膜。
(二)液态样品夹片法:适用于挥发性不大的液态样品。将液态样品滴在一片空白KBr片上,盖上另一片。外侧放上环形纸垫。涂片法:黏度大的液体样品,可涂在一片上,不夹片。液体池法:装入液体池(溶剂无干扰)。(三)气体试样:采用气体吸收池。先将气体吸收池中气体抽出,冲入适当压力的(约50mmHg)被测样品测定。(四)、衰减全反射(ATR):将纤维、高分子聚合物等难粉碎的样品,均匀铺展在衰减全反射棱镜的地面上,使紧密紧密接触,测定光谱图。
光谱解析方法光谱解析又称光谱诊断。(一)样品的来源和性质1.来源、纯度、灰分来源可帮助估计样品及杂质的范围。纯度>98%,不符合要求需精制。有灰分则含无机物。2.物理化学常数沸点、熔点等----旁证。3.分子式分子式可提供许多结构信息,可计算不饱和度,可判断是否含双键、三键、是饱和还是芳香等,还可验证光谱解析的合理性。
不饱和度即分子结构中距离达到饱和所缺一价元素的“对”数。例:C7H7NO规律:=0链状饱和化合物一个双键或一个脂环=1,若含≥1一个叁键=2,结构中含叁键,≥2一个苯环=4,结构中含苯环,≥4(二)光谱解析的几种情况1.已知物对照法2.查对标准光谱法3.简单化合物根据光谱解析即可判定4.新发现化合物待定结构或未收载标准光谱,综合解析(元素分析,四大光谱)
光谱解析程序先特征区,后指纹区先最强峰,后次强峰先粗查,后细找先否定,后肯定一抓一组相关峰
红外分光光度计的检定性能指标:分辨率、波数准确度与重现性、T%或A准确度与重现性、100%基线平直度及杂散光等。(一)波数准确度:(1)波数准确度的允许范围:傅里叶变换红外光谱仪在3000cm-1附近的波数误差应不大于±5cm-1,在1000cm-1附近的波数误差应不大于±1cm-1(2)波数准确度检定方法1、以聚苯乙烯薄膜校正(常用)2、以液体池用液体茚校正
(二)波数重复性:对同一张聚苯乙烯膜扫描3-5次,分别比较几次扫描图谱中个吸收峰的波数。(三)分辨率:以聚苯乙烯膜检定。傅立叶变换红外光谱仪以2cm-1分辨率进行扫描,记录光谱图。在3110-2850cm-1范围内应能显示7个吸收峰,其中峰2851cm-1与谷2870cm-1的分辨率深度应不小于18%透光率;峰1583cm-1与谷1589cm-1之间的分辨率深度应不小于12%透光率。仪器的标称分辨率应不低于2cm-1。(四)100%基线平直度偏差应小于4%透光率。(五)噪声:在1000cm-1处波数连续扫描5分钟,最大噪声(峰-峰值)应小于1%透光率。
红外分光光度计的日常维护一、环境要求:温度15-30℃,相对湿度小于65%。适当通风换气,以避免积累过量的二氧化碳和有机溶剂蒸汽。供电电压与接地电阻应符合仪器说明书要求。二、样品测定仓重放置一些干燥剂,常用变色硅胶或分子筛。注意观察干燥剂的颜色变化,做到及时更换干燥剂。三、压片磨具等一些红外附件,使用完后应及时清洗或擦拭干净,晾干,保存在干燥剂中,避免生锈。
红外分光光度计测定操作注意事项1、背景补偿或空白校正双光束仪器的参比光路中应置相应的空白盐片、溶剂或糊剂等;单光束仪器(FT-IR)应进行背景扫描,在供试品图谱中扣除背景吸收,即得供试品光谱。2、采用压片法时,常采用溴化钾。如样品为盐酸盐,常采用氯化钾。所用的溴化钾或氯化钾应预先研细,过200目筛,在120℃干燥4小时后保存在干燥其中备用。如有结块,须重新干燥。3、供试品研磨应适度,以粒度2-5um为宜。供试品过度研磨时会导致晶格结构的破坏或晶型的转化。粒度不够细易引起光散射能量损失,使整个光谱基线倾斜,甚至严重变形。4、常见的外界干扰因素二氧化碳:2350cm-1,667cm-1;水汽:3900-3300cm-11800-1500cm-1;溶剂蒸汽:干涉条纹。结果判定:红外光谱在药品分析中,主要用于定性鉴别。定性鉴别时,一是比较供试品光谱与标准图谱(«药品红外光谱集»)的全谱谱形;二是比较供试品光谱与对照品平行操作所得光谱的全谱谱形。首先比较谱带的有和无,然后比较各谱带的相对强度。如果两者完全相同,通常可判定为同一物质;两者如果不相同,须考虑供试品是否存在多晶现象,供试品纯度及外界干扰等因素后,再下结论。
谢谢'
您可能关注的文档
- 会计科目与账户专题培训PPT课件.ppt
- 会计凭证的基本内容培训PPT课件.ppt
- 最新2017年房地产入职新员工销售基础知识培训PPT课件.ppt
- 企业现金流管理实务培训PPT课件.ppt
- 企业国际化经营战略管理培训PPT课件.ppt
- 人力资源供给分析与规划培训PPT课件.ppt
- 临床销售拓展技巧培训PPT课件.ppt
- 上海市行政事业单位资产清查报表管理系统培训PPT ppt课件.ppt
- 桥梁荷载试验(广东省站上岗证培训PPT)ppt课件.ppt
- 2019年 柴油发动机培训PPT课件.ppt
- 2019 货架管理 卖场培训PPT课件.ppt
- 2016建筑行业营业税改增值税专题培训PPT课件.ppt
- 2015年幼儿园安全培训PPT.ppt
- 联轴器找中心培训PPT.ppt
- 组织结构设计培训PPT课件.ppt
- 白兰地和干邑酒水培训PPT.ppt
- 八大高危作业培训PPT.docx
- 职业安全与健康管理培训PPT.pptx