

- 12.03 MB
- 2022-04-29 14:44:55 发布

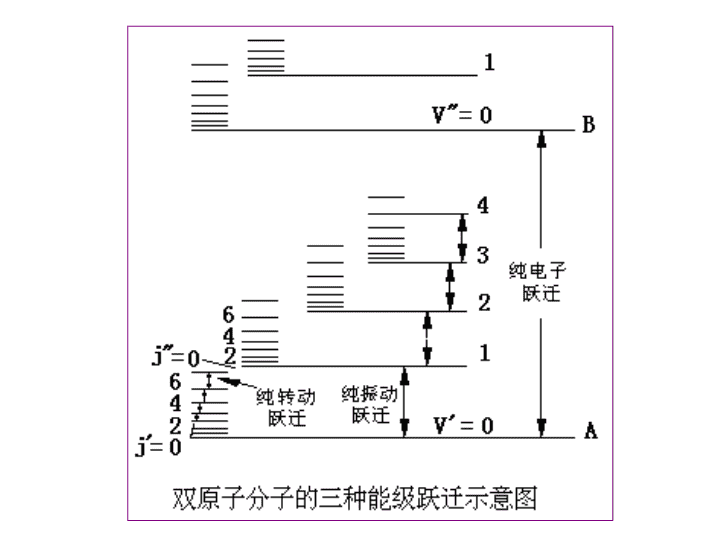
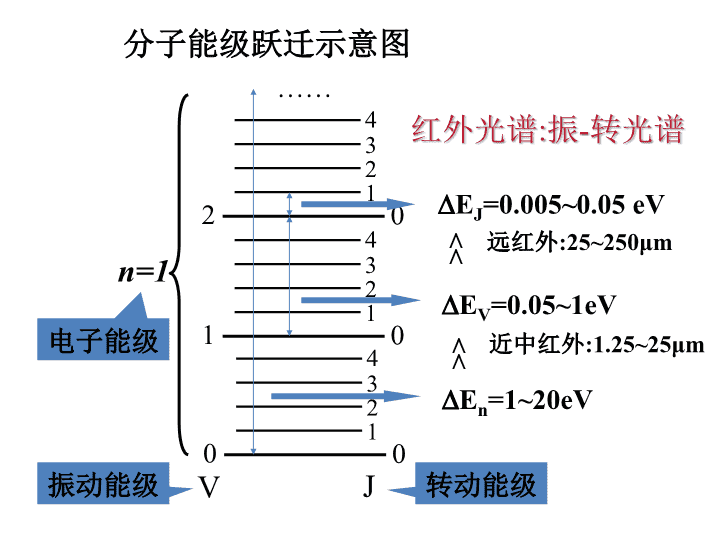
- 1、本文档共5页,可阅读全部内容。
- 2、本文档内容版权归属内容提供方,所产生的收益全部归内容提供方所有。如果您对本文有版权争议,可选择认领,认领后既往收益都归您。
- 3、本文档由用户上传,本站不保证质量和数量令人满意,可能有诸多瑕疵,付费之前,请仔细先通过免费阅读内容等途径辨别内容交易风险。如存在严重挂羊头卖狗肉之情形,可联系本站下载客服投诉处理。
- 文档侵权举报电话:19940600175。
'第三章红外光谱法
分子能级跃迁示意图012340112340212340……n=1VJ电子能级振动能级转动能级En=1~20eVEV=0.05~1eVEJ=0.005~0.05eV红外光谱:振-转光谱<<<<远红外:25~250μm近中红外:1.25~25μm
所以分子的能量E等于下列三项之和:E=Ee+Ev+Er式中Ee,Ev,Er分别代表电子能、振动能和转动能。分子从外界吸收能量后,就能引起分子能级的跃迁,即从基态能级跃迁到激发态能级。分子吸收能量具有量子化的特征,即分子只能吸收等于二个能级之差的能量:△E=E2-E1=hν=hc/λ
3.1红外吸收光谱分析概述红外吸收光谱又称为分子振动转动光谱。
红外光谱区分类名称λ/μmσ/cm-1能级跃迁类型近红外(泛频区)0.78~2.512820~4000OH,NH及CH键的倍频吸收中红外(基本振动区)2.5~254000~400分子中基团振动、分子振动远红外(转动区)25~300400~33分子转动,晶格转动
红外区的光谱除用波长λ表征外,更常用波数(wavenumber)σ表征。波数是波长的倒数,表示每厘米长光波中波的数目。若波长以μm为单位,波数的单位为cm-1,则波数与波长的关系是:
例如λ=5μm的红外线,它的波数为:
所有的标准红外光谱图中都标有波数和波长两种刻度(参见图10-3)。
3.2红外吸收光谱的产生条件红外光谱是由于分子振动能级的跃迁(同时伴随转动能级跃迁)而产生的。物质吸收电磁辐射应满足两个条件,即:(1)辐射应具有刚好能满足物质跃迁时所需的能量;(2)辐射与物质之回有偶合(coupling)作用(相互作用)。
红外辐射具有适合的能量,能导致振动跃迁的产生。当一定频率(一定能量)的红外光照射分子时,如果分子中某个基团的振动频率和外界红外辐射的频率一致,就满足了第一个条件。
为满足第二个条件,分子必须有偶极矩的改变。通常可用分子的偶极矩(dipolemoment)产来描述分子极性的大小。设正负电中心的电荷分别为+q和-q,正负电荷中心距离为d,则μ=q·d
并非所有的振动都会产生红外吸收,只有发生偶极矩变化的振动才能引起可观测的红外吸收谱带,我们称这种振动为红外活性(infraredactive)的,反之则称为非红外活性的(infraredinactive)。
3.3分子的振动光谱及方程式3.3.1谐振子分子中的原子以平衡点为中心,以非常小的振幅(与原子核之间的距离相比)作周期性的振动,即所谓简谐振动。这种分子振动的模型,用经典的方法可以看作是两端联接着的刚性小球的体系。最简单的例子是双原子分子,可用一个弹簧两端联着两个刚性小球来模拟,如图下所示。
双原子分子振动时,两个原子各自的位移如图所示。图中,re为平衡时两原子之间的距离,r为某瞬间两原子因振动所达到的距离。按照虎克定律,恢复到平衡位置的力F应与r—re成正比,即
(3-1)式中,K——化学键的力常数;δx1、δx2——分别为原子1、2在x轴上的位移;q——振动坐标。
由(3-1)(3-2)对于原子1(3-3)
v
对于原子2(3-4)
若只讨论重心不变的振动,有
(3-5)
联合(3-2)和(3-5),得
(3-6)
(3-7)
将(3-6)代入(3-3),再与(2-1)联合,可得
(3-8)
令或(3-9)μ为折合质量。再将(3-9)及(3-2)代入(3-8),得
(3-10)
解此微分方程得
(3-11)
式中q0——常数,代表振幅;λ——振动频率。
将(3-11)对t微分2次后代入(3-10),得
(3-12)
或用波数表示(3-13)若力常数K以N·m-1为单位,折合质量μ以原子质量单位为单位,则(3-13)可简化为(3-14)
例如,HCl分子的键力常数K为4.8×102N·m-1。根据(3—14)式可计算出HCl的振动频率为2892.4cm-1。σ=130.2
C-CC≡CC=CC-H>>>>同类原子→u"相同→K↑→↑不同原子→K起主导作用→K↑→↑u"起主导作用→u"↑→↓不同原子→K相近→u"↑→↓
上述计算值与实验值是很接近的。由计算可说明,同类原子组成的化学键(折合相对原子质量相同),力常数大的,基本振动频率就大。
例如分子中C—H键伸缩振动频率,该键力常数K=5×102N·m-1。实测值为2915cm-1,
几种基团伸缩振动频率的理论计算值与实际值
而真实分子的振动能量的变化是量子化的。
根据量子力学,该体系能量的薛定谔方程为
(3-15)
解为
(υ=0、1、2…)(3-16)
式中υ——振动量子数;Eυ——振动量子数υ相对应的体系能量。
利用(3-12),可将(3-16)式写成
(υ=0、1、2…)(3-17)
从(3-17)式可以看出,当υ=0时,体系能量仍不为0,此时能量为零点能。
从基态υ=0跃迁到第一激发态υ=1,2能级能量差ΔEυ为hν。按照产生吸收光谱的理论,吸收频率ν必须满足ν=ΔEυ/h,即双原子分子发生此跃迁时吸收光谱
的频率为,或,此结论与经典力学的结论相同。
3.3.2非谐振子双原子分子的实际位能随着核间距离的增大而增大,当核间距离增大到一定程度后,核间引力不再存在,位能为一常数。其位能曲线如图中实线所示。因此,位能函数应进行非谐振性的修正,为…(3-18)即非谐振子的位能应在谐振子的位能公式中加入校正项,通常只取到第二项,式中的x称为非谐振系数,其值远小于1。
真实分子具有非谐振性,相邻振动能级间能量差值ΔEυ并不相等,而是随着振动能级的能量升高而逐渐减少,但相邻的几个能级间的ΔEυ仍可视为相等。
3.6分子振动的形式设分子由n个原子组成,每个原子在空间都有三个自由度,原子在空间的位置可以用直角坐标系中的三个坐标x,y,z表示,因此n个原子组成的分子总共应有3n个自由度,亦即3n种运动状态。
振动形式应有(3n-6)种。
直线型分子的振动形式为(3n-5)种(图10-5)。
每一个振动自由度对应于一个基本振动,这些基本振动称为简正振动。下面举例说明之。
例1 水分子asOH3756cm-1sOH3652cm-1OH1595cm-1
例2二氧化碳分子asC=O2349cm-1弯曲振动666cm-1
亚甲基(-CH2-)的几种基本振动形式及红外吸收如图所示。
因此,分子的振动形式可分成两类。(l)伸缩振动(stretchingvibration)
i.对称伸缩振动(symmetricalstretchingvibration,σs);
ii.反对称伸缩振动(asymmetricalstretchingvibration,σas);
(2)变形或弯曲振动(deformationvibration)
i.面内变形振动(in-planebendingvibration,δ);剪式振动(scissoringvibration,δ);面内摇摆振动(rockingvibration,ρ);
ii.面外变形振动(out-of-planebendingvibration,γ);面外摇摆振动(waggingvibration,ω);扭曲变形振动(twistingvibration,τ)。
实际上,反映在红外光谱中的吸收峰有时会增多或减少,增减的原因为:(1)倍频谱带(overtoneband):合频谱带、差频谱带--泛频谱带。
泛倍频峰二倍频峰(ν=0→ν=2)频三倍频峰(ν=0→ν=3)峰(组)合频峰差频峰(即ν=1→ν=2,3…产生的峰)泛频峰强度较弱,难辨认→却增加了光谱特征性
(2)已知前述,并不是所有的分子振动形式都能在红外区中观察到。(3)有的振动形式虽不同,但它们的振动频率相等,因而产生简并(如前述CO2的面内及面外弯曲振动)。(4)费米共振和偶合共振(5)仪器分辨率不高,对一些频率很接近的吸收峰分不开。一些较弱的峰,可能由于仪器灵敏度不够而检测不出,等等。
3.7红外光谱的吸收强度根据量子理论,红外光谱的强度与分子振动时偶极矩变化的平方成正比。最典型的例子是C=O基和C=C基。醋酸丙烯酯的红外光谱C=O1745cm-1C=C1650cm-1
对于同一类型的化学键,偶极矩的变化与结构的对称性有关。例如C=C双键在下述三种结构中,吸收强度的差别就非常明显:(1)R-CH=CH2ε=40L·mol-1·cm-1(2)R-CH=CH-R’顺式ε=10L·mol-1·cm-1(3)R-CH=CH-R’反式ε=2L·mol-1·cm-1这是由于对于C=C双键来说,结构(1)的对称性最差,因此吸收较强,而结构(3)的对称性相对来说最高,故吸收最弱。
另外,对于同一试样,在不同的溶剂中,或在同一溶剂中不同浓度的试样中,由于氢键的影响以及氢键强弱的不同,使原子间的距离增大,偶极矩变化增大,吸收增强。谱带的强度还与振动形式有关。
应该指出,即使是强极性基团的红外振动吸收带,其强度也要比紫外及可见光区最强的电子跃迁小二到三个数量级。
另一方面,由于红外分光光度计中能量较低,使同一物质的摩尔吸收系数,随不同仪器而改变。这就使在定性鉴定中用处不大。摩尔吸光系数谱带强度表示符号>100非常强峰vs20~100强峰s10~20中等强度峰m1~10弱峰w<1非常弱峰vw
3.8影响基团频率位移的因素外部因素内部因素电子效应空间效应氢键效应杂化影响互变异构费米共振溶剂物态晶型仪器
3.8.1外部因素(1)物态效应正己酸的气态(a)和液态(b)红外吸收光谱
奥克托金三种晶性型的红外光谱
在极性溶剂中,溶质的极性基团的伸缩振动频率,往往随着溶剂的极性增加而降低,但吸收强度增加。例如,对-二甲胺基硝在CCl4和CCl3CN两种不同溶剂中的硝基σas分别为1506cm-l和1496cm-1。σs分别为1332cm-l和1320cm-l。
此外,溶液的浓度和温度的变化也能引起谱带的变化。
(2)溶剂效应:极性↑→↓原因:形成分子间氢键化合物溶剂伸缩振动频率(cm-1)甲醇OHCCl43644Et2O3508N(CH2CH3)33243丙酮C=OC6H141727CCl41720CHCl3、CHBr3、CH3CN1705红外光谱常用溶剂:CCl4、CS2、氯仿、二氯甲烷、乙腈、丙酮等,T=20~60%。
3.8.2内部效应(1)电效应(electricaleffects)由子化学键的电子分布不均匀而引起。i.诱导效应(inductiveeffect)由于取代基具有不同的电负性,通过静电诱导作用,引起分子中电子分布的变化,从而引起键力常数的变化,改变了基团的特殊频率,这种效应通常称作诱导效应。现从以下几个化合物来看诱导效应(直箭头表示)引起C=O频率升高的原因。
R-CORC=01715cm-1;R-COHC=01730cm-1;R-COClC=01800cm-1;R-COFC=01920cm-1;F-COFC=01920cm-1;R-CONH2C=01928cm-1;1715cm-11800cm-11828cm-11928cm-1
ii.共轭效应(conjuqativeeffect)
共轭效应使共扼体系中的电子云密度平均化,结果使原来的双键伸长(即电子云密度降低),力常数减小,所以振动频率降低。例如酮的C=O,因与苯环共扼而使C=O的力常数减小,频率降低:cm-1cm-1cm-1cm-1
当含有孤对电子的原子接在具有多重键的原子上时,也可起类似的共轭作用。例如在这化合物中,由于N原子的吸电子作用存在诱导效应,但比共轭效应影响小,因此C=O的频率与饱和酮相比还是有所降低,这是I效应与M效应同时存在的例子之一。1650cm-11735cm-1
I效应与C效应同时存在的例子还有饱和酯。饱和酯的C=O伸缩频率为1735cm-1,比酮(1715cm-1)高,这是因为-OR基的I效应比M效应大,所以C=O的频率升高。-I<+C-I>+C
iii.偶极场效应(dipolarfieldeffect)偶极场效应(F效应)要经过分子内的空间才能起作用,因此相互靠近的官能团之间,才能产生F效应。如氯代丙酮有三种旋转异构体:
(2)氢键(hydrogenbonding)
游离羧酸的C=O频率出现在1760cm-1左右,而在液态或固态时,C=0频率都在1700cm-1左右,因为此时羧酸形成二聚体形式。氢键使电子云密度平均化,C=0的双键性减小,因此C=0的频率下降。
乙醇在不同浓度CCl4中的部分红外光谱
(3)振动的偶合(vibrationalcoupling)
适当结合的两个振动基团,若原来的振动频率很相近,它们之间可能会产生相互作用而使谱峰裂分成两个,一个高于正常频率,一个低于正常频率。这种两个振动基团之间的相互作用,称为振动的偶合。例如酸酐的两个羧基,振动偶合而裂分成两个谱峰:~1820cm-1~1760cm-1
此外,二元酸的两个羧基之间只有1-2个碳原子时,会出现两个C=0吸收峰,这也是由偶合产生的:
(4)费米共振(Fermiresonance)
当一振动的倍频与另一振动的基频接近时,由于发生相互作用而产生很强的吸收峰或发生裂分,这个现象叫做费米共振。例如醛基的C-H伸缩振动基频与弯曲振动的倍频发生费米共振,在2820cm-1和2720cm-1两处出现两个吸收峰,这两个吸收峰上醛类化合物的特征吸收峰。
(5)立体障碍(stericinhibition)
由于立体障碍,羰基与双键之间的共轭受到限制时,σC=O较高,例如:在(II)中由于接在C=O上的CH3的立体障碍,C=O与苯环的双键不能处在同一平面,结果共轭受到限制,因此σC=O比(I)稍高。
(6)环的张力(ringstrain)
环的张力越大,σC=O就越高。在下面几个酮中,4元环的张力最大,因此它的σC=O最高。
环张力效应使得环外双键的伸缩振动频率升高,而环内双键的伸缩振动频率降低。
CHCHCHCH1576cm-11611cm-11644cm-11781cm-11678cm-11657cm-11651cm-1CH3060-3030cm-12900-2800cm-12222
互变异构杂化影响(hybridizationaffect)不饱和氢CH>3000cm-1;饱和氢CH<3000cm-1s成分↑→CH↑
10.6红外光谱的特征性,基团频率红外光谱的最大特点是具有特征性。复杂分子中存在许多原子基团,各个原子基团(化学键)在分子被激发后,都会产生特征的振动。分子的振动,实质上可归结为化学键的振动。
(1)X-H伸缩振动区:4000-2500cm-1。X可以是O,H,C和S原子。在这个区域内主要包括O-H,N-H,C-H和S-H键伸缩振动,通常又称为“氢键区”。
(2)叁键和累积双键区:2500-1900cm-1。
主要包括炔键-C≡C-,腈键-C≡N、丙二烯基-C=C=C-、烯酮基-C=C=O、异氰酸酯基-N=C=O等的反对称伸缩振动。
(3)双键伸缩振动区:1900-1200cm-1。
主要包括C=C,C=O,C=N,-NO2等的伸缩振动,芳环的骨架振动(skeletalvibration)等。
(4)X-Y伸缩振动及X-H变形振动区(单键区)(含指纹区):<1650cm-1。
这个区域的光谱比较复杂,主要包括C-H,N-H变形振动,C-O,C-X(卤素)等伸缩振动,以及C-C单键骨架振动等。
10.6.1烷烃烷烃化合物含有CH3、CH2和CH等基团,其红外吸收带主要由C—H键和C—C键的振动引起。见下图。正庚烷的红外光谱(CH3(CH2)5CH3)
(1)C-H键不对称和对称分别在2960cm-1和2870cm-1附近。一般σas>σs。当分子中同时存在CH3和CH2时,C-H键在3000~2800cm-1附近间一般有4个吸收峰。
(2)C-H键不对称和对称弯曲振动分别在1470cm-1和1380cm-1附近。a孤立CH3在1380cm-1附近出现单峰,CH3越多,峰强越强;b偕二甲基(=C(CH3)2)的2个CH3对称弯曲振动偶合,裂分为双峰,在1385cm-1和1375cm-1附近,强度几乎相等。c叔丁基(-C(CH3)3)的3个CH3对称弯曲振动偶合,裂分为双峰,在1395cm-1和1370cm-1附近,强度后者是前者的2倍,以此与(=C(CH3)2)相区别。
d当CH3与杂原子相连时,1380cm-1附近峰由于诱导效应发生位移,相邻基团的电负性越大,1380cm-1峰越向高波数移动。-O-CH3=N-CH3≡C-CH3≡Si-CH3~1440cm-1~1426cm-1~1380cm-1~1255cm-1
(3)亚甲基CH2的平面摇摆振动。当分子中含有-(CH2)n-(n≥4)时,在725cm-1有峰,并随着n的减少,逐渐向高波数移动。
(4)CH3和CH2的区别在于:CH3在1380cm-1附近有吸收峰,而CH2没有。
(5)C-C键的骨架振动在1250~800cm-1附近,因特征性不强,用处不大。
10.6.2烯烃
(1)烯烃不饱和C原子上的C-H伸缩振动在3100~3000cm-1,强度中等,峰形较尖锐。末端烯烃(RCH=CH2、R1R2C=CH2)的σ=CH的吸收频率较高,在3095~3075cm-1有强峰,而其它取代乙烯的σ=CH在3040~3000cm-1。小环烷烃、环氧及环氮化合物,环上的C-H键的伸缩振动频率也在3000cm-1以上。
(2)C=C的伸缩振动频率在1680~1620cm-1:基团伸缩振动频率(cm-1)强度R-CH=CH21645中强R2CH=CH21655↓(顺)RCH=CHR1660↓(反)RCH=CHR1675↓三取代或四取代物1670弱
(3)烯烃不饱和碳原子上的C-H面外弯曲振动在1000~650cm-1,对结构敏感:一取代双键(RCH=CH2)995~985cm-1(m),915~905cm-1(s)
倍频1860~1790cm-1α,α-二取代(RR’C=CH2)895~885cm-1(s),倍频1860~1750cm-1反式二取代(-CH=CH-)980~960cm-1(s)顺式二取代(-CH=CH-)730~665cm-1(s)三取代(RR’C=CHR”)850~790cm-1(m)
10.6.3炔烃1-辛炔的红外光谱
一元取代炔烃的红外吸收光谱有三个特征吸收带:(1)炔烃的σ≡CH在3300cm-1附近,峰形尖锐,容易识别。(2)炔烃的σC≡C在2140~2100cm-1,一般强度较弱。炔烃的烷基二取代物中,σC≡C在2260~2190cm-1,由于分子的偶极矩变化小,一般难以观察到。(3)炔烃的面外C≡C在700~600cm-1,吸收带强而较宽。
10.6.4芳烃1—芳环的CH;2—芳环的C=C
3—芳环的面外=CH;4—芳香醚的不对称C-O-C
芳烃的红外光谱主要有三个特征吸收带:(1)芳环上的C-H键的伸缩振动频率在3100~3000cm-1。(2)芳环的骨架振动频率σC=C一般有四个谱带:1600、1585、1500、1450cm-1附近。其中1600和1500cm-1是芳环的特征吸收带,其强度后者比前者强。(3)芳环上的面外弯曲振动σ=CH(即取代)
苯670cm-1(s)
一元取代770~730cm-1(s),710~690cm-1(s)
二元取代(邻位)770~735cm-1(s)
二元取代(间位)900~860cm-1(m),810~750cm-1(s)725~680cm-1(m)
二元取代(对位)860~800cm-1(s)
三元取代(1,3,5取代)860~810cm-1(m),735~675cm-1(s)
三元取代(1,2,3取代)780~760cm-1(m),725~680cm-1(s)
三元取代(1,2,4取代)885~870cm-1(m),825~805cm-1(s)
四、五元取代900~800cm-1(s)
σ=CH在2000〜1650cm-1范围出现的泛频吸收(有机物结构中无羰基=CO)
10.6.5醇、酚α-辛醇的红外光谱(液膜0.025mm)
乙醇在不同浓度CCl4中的红外谱图
醇和酚的O-H键及C-O键的伸缩振动吸收在红外光谱中是鉴定它们的特征频带:(1)醇和酚通常以二缔合或多缔合态存在。缔和态的OH伸缩振动频率在3550~3200cm-1,谱带强而宽。若为二缔合态,吸收带的中心在3500cm-1附近,多缔合态一般在3320cm-1附近。游离态σOH在3650~3590cm-1,谱带尖锐,强度中等。结晶水的σOH也在3600~3000cm-1,但一般强度低而且峰窄。水峰同时在1670~1600cm-1附近出现H-O-H的弯曲振动峰,以示区别。
(2)醇和酚的C-O伸缩振动频率在1260~1000cm-1(s)。因OH连接的碳原子类型不同:
伯醇:1050cm-1左右
仲醇:1100cm-1左右
叔醇:1150cm-1左右
酚:1200cm-1左右
(3)醇的O-H键面内弯曲振动频率在1420~1330cm-1(同CH2或CH面外弯曲振动频率区)。酚的面外弯曲振动频率在1350cm-1左右,较宽,强度低于C-O伸缩振动频率吸收带。
10.6.6醚
醚的红外特征吸收带是C-O-C键的不对称伸缩振动:
脂肪醚1150~1060cm-1(s)
芳香醚1275~1210cm-1(s)
乙烯基醚1225~1200cm-1(s)
在这一区域附近,醇、醛、酮、酯和内酯等均有红外吸收。因此,在红外光谱中只有不存在羟基和羰基的其它特征峰,而在此区域有吸收峰时,才说明被测分子中有醚基存在。
10.6.7羰基化合物羰基伸缩振动频率(cm-1)酸酐18201760酰卤1800酯1740醛1730酮1715羧酸1700酰胺1690
10.6.7.1醛正丁醛的红外光谱(液膜,0.025mm)1—醛基中的CH;2—C=O
醛基有CHO两个特征吸收峰:
(1)羰基的C=O伸缩振动在1740~1720cm-1(s);
(2)醛基中的CH伸缩振动基频与CH弯曲振动的倍频发生费米共振,在2820和2720cm-1出现2个窄的中等强度的吸收峰,高波数的吸收峰常被饱和次甲基的CH伸缩振动吸收峰覆盖,因此常常只观察到2720cm-1的吸收峰。这是鉴定醛基的依据。
10.6.7.2酮酮类的羰基伸缩振动吸收带是其唯一的特征吸收带。3,3-二甲基-2-丁酮的红外光谱
10.6.7.3羧酸和羧酸盐1-羧酸二缔合态的OH伸缩振动;2-C=O的伸缩振动;3-羧酸二缔合态的OH面外摇摆
在羧酸中,羧基COOH的O-H键伸缩振动、C一O键伸缩振动以及O-H键弯曲振动噘收是红外光谱中识别羧酸的主要特征峰:
(1)羧酸通常以而缔合态存在。缔合态的OH伸缩振动频率在3200~2500cm-1,峰较强,峰形宽而散。这个谱带在2700~2500cm-1之间通常出现几个连续小峰,很特征。在稀溶液或非极性溶剂中羧酸为游离态,OH伸缩振动频率在3550cm-1附近,为强吸收峰。
(2)羧酸的C=O伸缩振动频率缔合时在1725~1700cm-1,游离态在1760cm-1附近。
(3)羧酸二缔合态的OH面外弯曲振动频率在955~915cm-1,为一个宽而不强的吸收峰,常在羧酸的鉴定中作参考。
(4)羧酸盐中,羧酸根负离子COO-的2个CO是均等的,其不对称和对称伸缩振动频率分别在1610~1550cm-1(S)和1420~1300cm-1(m)。
10.6.7.4酯
在酯基中(COOC),C=O及C-O-C伸缩振动吸收是红外光谱中酯类的2个特征吸收峰:
(1)绝大多数饱和羧酸酯的C=O伸缩振动都位于1740cm-1附近;
(2)酯基中的C-O-C伸缩振动在1300~1000cm-1有2个吸收峰,常为酯类化合物的最强吸收峰。其中C-O-C不对称伸缩振动在1300~1150cm-1(S),对称伸缩振动在1140~1000cm-1(w),是鉴定酯类的重要依据。(3)在酯类中,有时可在3450cm-1附近看到C=O的倍频吸收峰。
10.6.7.5酸酐乙酸酐的红外光谱图C=O伸缩振动1828、1750cm-1;C-O-C伸缩振动1125cm-1
酸酐的C=O及C-O-C伸缩振动吸收是红外光谱中酯类的2个特征吸收峰:
(1)酸酐的C=O伸缩振动由于分子中的2个羰基伸缩振动偶合,在1860~1800cm-1和1800~1750cm-1间出现2个吸收峰,分别是羰基的不对称和对称伸缩振动,相距60cm-1左右。开链酸酐,高频率比低频率略强;环状酸酐,低频率比高频率略强。这是酸酐类化合物的特征谱带。
(2)酸酐的C-O-C伸缩振动为强而宽的吸收峰,开链酸酐在1170~1050cm-1,而环状酸酐在1310~1200cm-1。
10.6.7.6酰卤苯甲酰氯的红外光谱1-C=O伸缩振动;2-2×871cm-1因费米共振强度增加;3-C-C
酰卤中由于卤原子与羰基相连,C=O伸缩振动移至1800cm-1附近。
在芳香酰卤中,由于875cm-1附近的C-C伸缩振动的倍频与羰基伸缩振动发生费米共振,所以羰基吸收范围内出现双峰,通常高频率较强。
10.6.7.7酰胺甲酰胺的红外谱图1-NH伸缩振动;2-C=O伸缩振动(I带)
3-NH(II带)
酰胺的红外谱图有3个主要的特征峰:
(1)酰胺的NH伸缩振动在3000cm-1以上的高频区:伯酰胺的NH伸缩振动是不对称和对称振动的双峰(m)。在稀溶液中在3500cm-1和3400cm-1附近;在液态或固态时,分子间缔合使其移动到3350cm-1和3180cm-1附近。仲酰胺的NH伸缩振动为单峰,在稀溶液中3460~3400cm-1间为一个峰。由于C-N的部分双键性质引起顺式和反式2种异构体,使其分裂为很靠近的双峰(高分辨率仪器)。液态或固态时,峰在3300cm-1间处。叔酰胺无此峰,可辨别伯、仲、叔酰胺。
(2)酰胺的C=O键伸缩振动吸收峰常称为为I带,略低于相应的酮:
伯酰胺在1690cm-1附近;
仲酰胺在1680cm-1附近;
叔酰胺在1670~1630cm-1。缔合态时,三者都在1650cm-1附近。
(3)酰胺的NH面外弯曲振动常称为酰胺的II带。
伯酰胺在比C=O低的频率出现尖峰,其强度相当于C=O峰强的1/3到1/2。在稀溶液中,伯酰胺的NH在1620~1590cm-1;固态时升至1650~1620cm-1。缔合态的II带常与I带重叠为1个吸收峰。;
仲酰胺在稀溶液中在1550~1510cm-1,在固态时移至1570~1515cm-1。仲酰胺的I带和II带能够清晰分开。因此会区分伯、仲酰胺。
叔酰胺无酰胺II带。
10.6.8胺和胺盐叔丁基胺的红外光谱(液体,池厚0.01mm)
l-不对称NH伸缩振动;2-对称NH伸缩振动;3-NH(面内);4—C-N
(1)伯胺的NH伸缩振动,在稀溶液中在3500~3300cm-1处出现2个吸收峰(不对称和对称)。脂肪族较弱;芳香族可达中等。
仲胺稀溶液在此区域只出现1个峰,脂肪族很弱,常观察不到;芳香族较强。当分子中同时NH和OH时,吸收峰发生有差别的重叠:OH峰强而宽;NH峰弱而尖锐。
(2)伯胺的NH面内弯曲振动在1650~1580cm-1处,为中等强度的宽峰;
仲胺在此很弱。
(3)胺的C—N伸缩振动,只有芳胺为强峰。各类芳胺的位置不同:
伯芳胺在1340~1250cm-1处;
仲芳胺1350~1280cm-1处;
叔芳胺1360~1310cm-1处;
脂肪族胺的CN强度弱,结构测定中用处不大。
(4)铵盐的NH:
伯铵盐在3200~2800cm-1处出现NH3+的二个较宽的强吸收带;
仲铵盐在3000~2700cm-1处出现NH2+的较宽强吸收带;
叔铵盐在2700~2330cm-1处出现NH+吸收带。
此外,伯铵盐还可看到NH3+的不对称和对称伸缩振动在1650~1560cm-1及1550~1505cm-1。仲铵盐的NH2+弯曲振动在1620~1560cm-1。
10.6.9硝基化合物硝基化合物中硝基NO2的不对称和对称伸缩振动吸收是其红外特征吸收带。
10.6.9.1脂肪族硝基化合物(1)脂肪族的硝基化合物的NO2的不对称和对称伸缩振动分别在1560和1350cm-1附近一般都较强,通常不对称伸缩振动更强。(2)硝基甲烷有较高的硝基不对称伸缩振动频率。随着硝基邻接的碳原子上的氢原子数目的减少,吸收峰逐渐略微降低。(3)CN通常在920~800cm-1处(M)。
10.6.9.2芳香族硝基化合物芳香族硝基化合物的NO2的不对称和对称伸缩振动比脂肪族低约20~40cm-1,两峰强度相似,有的甚至对称伸缩振动峰更强。
10.6.10腈腈类化合物中,C≡N的伸缩振动峰是其特征峰:饱和脂肪腈化合物,C≡N的伸缩振动频率在2260~2240cm-1(m)。不饱和腈类化合物的C≡N伸缩振动频率在2240~2225cm–1,为强谱带。芳香腈化合物在2240~2225cm-1处有一个较脂肪腈的谱带要强的C≡N伸缩振动峰,其谱带强度受取代基性质的影响很大。
10.6.11有机卤代物有机卤代物中,C-X键的伸缩振动峰是特征峰。
10.6.11.1有机氟代物有机氟代物的C-F伸缩振动频率在1400~1000cm–1(s):CF在1100~1000cm–1CF2在1280~1120cm–1,多重峰,(s)CF3在1350~1120cm–1,多重峰,(s)
10.6.11.2有机氯代物有机氯代物中的C-Cl键的伸缩振动在800~600cm–1。CH2-Cl基在760~700cm–1,CH-Cl基在640~610cm–1。在CCl4中,C-Cl在797cm–1。C-Cl键的伸缩振动吸收谱带的位置与分子的构象有关。
10.6.11.3有机溴代物有机溴代物中的C—Br键伸缩振动在700~500cm–1。反式构象在650cm–1附近;顺式构象在560cm–1附近。在甾族化合物中,C—Br平伏键在750~700cm–1;直立键在690~590cm–1。
10.6.11.4有机碘代物有机碘代物中的C—I伸缩振动在600~500cm–1。反式构象在600cm–1附近;顺式构象在500cm–1附近。
10.6.12有机硫化物含巯基S—H的有机物,其S—H键伸缩振动频率在2590~2550cm–1,为弱吸收谱带。因S—H基形成氢键的倾向很小,在液态和稀溶液中该吸收带位置变化不大。C—S键伸缩振动吸收频率在705~570cm–1,谱带强度也很弱。凡含有S=O和S=O=S的化合物,S=O的伸缩振动产生强吸收峰。例如砜和亚砜类物质。
10.7仪器和实验10.7.1红外光谱仪10.7.1.1色散型红外吸收光谱仪目前常用的红外光谱仪为色散型红外吸收光谱仪。它的构造基本上由光源、单色器、样品池、检测器、放大记录器等五个部分组成。
色散型红外光谱仪结构示意图
10.7.2样品处理技术10.7.2.1气态样品气体或低沸点的液体样品,可以直接导入已抽成真空的气体池内测定。气体池的主体是一个玻璃筒,两端为氯化钠或溴化钾晶片,池内的反射镜使红外光多次反射以大大增加光程。
10.7.2.2液体样品(1)纯液体样品可直接注入光程长度为0.01~1mm的液体吸收池内进行测量,这就是谱I常注明的液膜法。所谓液膜法即在两块圆形岩盐片之间滴1~2滴液体样品,形成很薄的膜,用专用夹具将两块盐片夹住便可进行测量。在两块盐片之间垫入不同厚度的垫片,可以节液膜的厚度。
(2)稀溶液样品的光谱可以提高数据的重复性。通过选择适当的浓度(0.1~10%)和光程长度(0.01~1mm),可以使某些重要基团的吸收谱带清晰地显示出来。但是,由于溶剂本身也有红外吸收,所以一次不可能得到一张完整的红外光谱。常用于红外测定的溶剂的透明范围见表2-1。
10.7.2.3固体样品(1)KBr压片法这是测定固体样品最常用的方法。一般取l~2mg样品粉末和100mg左右干燥的KBr粉末一起放入玛瑙研钵中研细混匀,然后倒入专用的压片器中,一边抽真空,一边加压,制成透明的薄圆片。将此片放入仪器的样品架上便可进行测量。此法因KBr吸潮,常在3500cm-1及1640cm-1附近出现水的干扰吸收峰。纯KBr在4000~400cm-1无吸收,因此,压片法可获得样品的红外全图谱。
(2)研糊法
将2~5mg样品充分研细成小于2μm的细小颗粒,滴入l~2滴石蜡油作分散剂进一步研磨,直至呈均匀糊状物。然后将研糊物放在两盐片之间,压成一薄层物进行测量。也可以用六氯丁二烯或全氟煤油等调糊,它们没有碳一氢振动吸收的干扰。
对于可溶解的高聚物样品,可以将其溶液倒在玻璃板上,待溶剂挥发后,直接用高聚物的膜进行测定。
10.7.3傅里叶变换红外光谱仪(简称FT—IR)
FT-IR主要是由光源、迈克尔逊干涉仪、探测器和计算机等部分组成。FTIR的核心部分是迈克尔逊干涉仪,图10-15是它的光学示意和工作原理图。
图10-16(b)为另一入射光波长为λ2的单色光所得干涉图。如果两种波长的光一起进入干涉仪,则将得到两种单色光干涉图的加合图(图10-16(c))。同样,当入射光为连续波长的多色光时,得到的则是具有中心极大并向两边迅速衰减的对称干涉图(图10-17)。这种多色光的干涉图等于所有各单色光干涉图的加合。若在此干涉光束中放置能吸收红外光的试样,如图10-14所示,由于试样吸收了某些频率的能量,结果所得到干涉图强度曲线函数就发生变化。
图10-16(a)所示的单色辐射的干涉图是一种余弦波,可用下述方程表示:式中,I(δ)是干涉图的强度,ν是波数,δ是光程差,B(ν)代表与仪器参数有关的光源的强度,即代表光源的光谱。同样对双色辐射如图10-16(b)所示,相应的干涉图方程为:
则对连续辐射如图10-16(c)所示,则干涉图为所有无数余弦项的加和即:
在数学上,上述方程中的I(δ)称之为谱B(ν)的余弦傅立叶变换。
如果要从干涉图计算光谱B(ν),即通过计算I(δ)的余弦傅立叶变换。因此这种光谱技术称之为傅立叶变换光谱法。对连续光源,上述方程的逆傅立叶变换为:或
FTIR仪器由于没有狭缝的限制。光通量只与干涉仪平面镜大小有关,因此在同样分辨率下,光通量要大得多,从而使检测器接受到的信号和信噪比增大,因此有很高的灵敏度,有利于弱光谱的测定;扫描速度极快,能在很短时间内(<1s)获得全频域光谱响应;由于采用激光干涉条纹准确测定光程差,使FTIR测定的波数更为准确等优点。
10.8红外光谱定性分析(1)用各种分离手段(如分馏、萃取、重结晶、层析…)提纯试样,以得到单一的纯物质。(2)了解与试样性质有关的其它方面的资料
不饱和度计算公式:U=1+n4+2n6+1/2(n3+3n5-n1)式中,n1-1价元素,包括+1价的碱金属和
-1价的卤素。例如H,Na,Cl;n2-2价元素,例如O;n3-3价元素,例如N;n4-4价元素,例如C;n5-5价元素,例如P,N;
n6-6价元素,例如S。
U=0,化合物饱和;U=1,化合物分子中一定有一个双键或一个饱和环状化合物(-NO2只算一个双键,所以CH3NO2的U=1);U=2,分子中一定有一个三键或两个双键……U=3,由上述情况判断;U=4,分子中含一个苯环或由上述情况判断。
(3)解析红外谱图法A区域法B缩小范围法(区分无机物和有机物)C主要官能团解析法:C=O、O-H、N-H、C-O、C=C、C≡C、C≡N、Ar、-NO2等
a判断C=O是否存在:
在1870~1660cm–1间有1(或2)个最强强吸收峰。
羧C=O是否存在酸、酰胺、酸酐、酯类、醛类(酰卤)、酮类b如果C=O不存在:
醇和酚、胺类、醚类c双键和芳环d三键e硝基f烷类
例1某未知物为C8H10
例2某未知物为C8H18O
例3某未知物为C10H12O
例4某未知物为C4H9O3N
例5某未知物为C9H8O4
例6某未知物为C8H10O
例7某未知物为C5H122985~2880cm-11460cm-1、1388cm-1
例8某未知物为C6H122930~2855cm-11453cm-1
例9某未知物为C9H103090~3035cm-12978~2921cm-11973~1750cm-11635cm-11605cm-11577cm-11498cm-11390cm-1888cm-1770cm-1700cm-1
例10某未知物为C8H143300cm-1<3000cm-12100cm-11470cm-11370cm-1720cm-1625cm-1
例11某未知物为C8H8O2
例12某未知物为C3H7Br
例13某未知物为C7H8O
例14某未知物为C4H11N
例15某未知物为烯丙醇的简单衍生物
例16某未知物为C4H10O
例17某未知物为C8H8O
例18某未知物为C8H10
例19某未知物为C3H6O
例20某未知物为C7H8O
例21某未知物为C8H7N
例22某未知物为C6H4FNO2
例23某未知物为C10H10O
例24某未知物为C8H8
例25某未知物分子量为53
例26某未知物为C8H8O
例27某未知物为C7H6O2
例28某未知物为C、H、N、S
例29某未知物为C7H6O
例30某未知物为C6H14
例31某未知物为C8H7N
例32某未知物为C4H6O2
例33某未知物为C10H14S
例34某未知物为C8H18
例35某未知物为C4H6O2
例36某未知物为C6H12O
例37一个含CHNO4种元素的固体化合物,熔点146度,有很好的水溶性,由红外谱图(石蜡糊法)推断其结构。
例38某未知物为C4H6O3N,红外光谱在3020、3010、2975、2850、1760、1585、1485、1450、1270cm-1等处有强度不同的吸收峰,试推断其结构。(陈洁)
10.9红外光谱定量分析和其它吸收光谱分析(紫外、可见光吸光光度法)一样,红外光谱定量分析是根据物质组分的吸收峰强度来进行的。它的依据是朗伯-比尔定律。各种气体、液体和固态物质,均可用红外光谱法进行定量分析。有关红外光谱定量分析的方法可参阅有关书籍。
随着计算机的发展和应用以及光谱仪器的进展,红外光谱定量分析亦得到发展。上述一些困难正在不断克服中。特别是多组分试样的定量分析,已有许多商品化的红外光谱定量分析软件包,如最小二乘法、修正矩阵法、因子(主因子)分析法、卡尔曼滤波法、神经网络法等,可同pc机兼容及相关的各类红外光谱仪连接。'
您可能关注的文档
- 幼儿园小班数学课件PPT_认识形状 (2).ppt
- 幼儿园小班数学课件PPT_认识形状 (3).ppt
- 高中生物说课课件PPT.doc
- 上课用_功的课件PPT-新版人教.ppt
- 人教版四年级语文下册《永生的眼睛》课件PPT.ppt
- 七年级1班家长会课件PPT.ppt
- 伯牙绝弦课件PPT.ppt
- 北师大版小学数学二年级下册《平行四边形》课件PPT[1].ppt
- 小学三年级数学分数的初步认识第1课时课件PPT2.ppt
- 建筑材料见证取样及送检课件PPT.ppt
- 最新苏教版五年级数学下册《约分》课件PPT.ppt
- 不等式的性质课件PPT25张(人教版七年级下数学).ppt
- 的乘法口诀课件PPT(人教新课标二年级上册数学课件)冯秀霞 .ppt
- 管理培训课件PPT(精).ppt
- 落日的幻觉课件PPT .ppt
- 《圆锥的认识》课件PPT hao.ppt
- 主题班会高一入学教育课件PPT课件.ppt
- 人教版二年级上册识字一课件PPT修改.ppt